
Istnieją trzy typy koronawirusa SARS-CoV-2

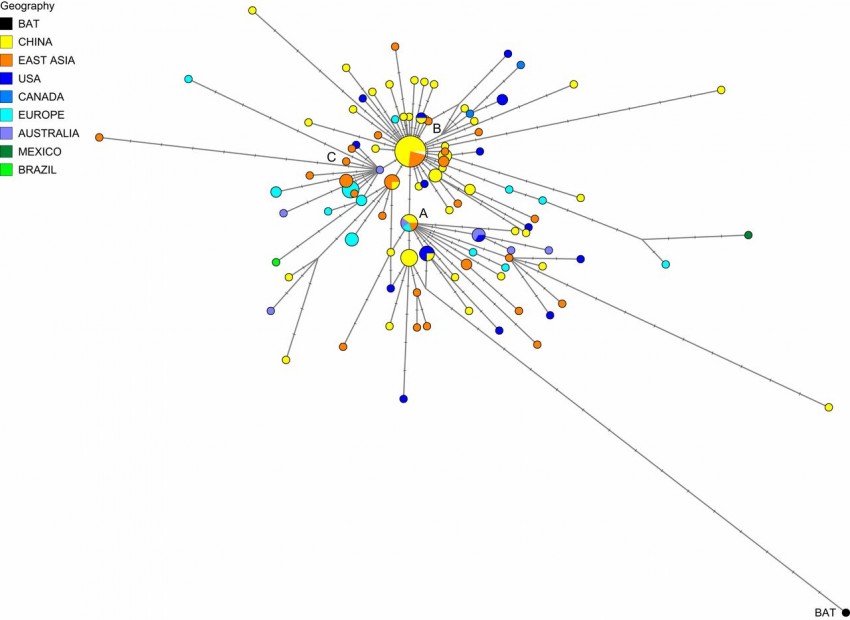
Niemiecko-brytyjski zespół specjalistów z University of Cambridge, Uniwersytetu Christiana Albrechta w Kilonii oraz Instytutu Genetyki Sądowej w Münster, wykonał analizę filogenetyczną koronawirusa SARS-CoV-2. Naukowcy wykorzystali przy tym genom wirusów pobranych od 160 pacjentów pomiędzy 24 grudnia 2019 roku a 4 marca 2020 roku. Analiza ujawniła istnienie trzech typów wirusa, które naukowcy nazwali A, B oraz C.
Typ A to jest najbliższy „oryginalnemu” koronawirusowi od nietoperzy. Typu A i C stanowią zdecydowaną większość wersji wirusa spotykanych poza Azją Wschodnią. Z kolei typ B występuje głównie w Azji Wschodniej, a wydostał się poza nią dopiero po przejściu mutacji.
Niespodzianek jest więcej. Na przykład typ A, ten, który jest najbliższy koronawirusowi nietoperzy, jest co prawda obecny w Wuhan, ale nie jest tam dominujący. Zmutowane wersje typu znaleziono u Amerykanów mieszkających w Wuhan oraz u wielu mieszkańców USA i Australii.
Z kolei typ C bardzo rozpowszechnił się w Europie. Zidentyfikowano go już u pierwszych pacjentów we Francji, Szwecji, Włoszech i Anglii. Brak go w próbkach z Chin kontynentalnych, jednak jest obecny w Hongkongu, Singapurze i Korei Południowej.
Z analizy wynika również, że do Włoch koronawirus mógł dostać się z Niemiec oraz z Singapuru.
Jak zapewniają autorzy badań, zastosowana przez nich metoda dokładnie pokazuje trasy rozprzestrzeniania się epidemii, poszczególne mutacje i typy wirusa są dobrze połączone. Dzięki temu, jak twierdzą, można wykorzystać ich metodę do zidentyfikowania nieznanych obecnych oraz przyszłych miejsc, z których choroba może się rozprzestrzeniać. Analiza sieci filogenetycznej może pomóc w zidentyfikowaniu nieznanych źródeł COVID-19, co pozwoli na objęcie ich kwarantanną, mówi doktor Peter Foster, główny autor badań.
Wiemy zatem, że wariant A jest najbliżej spokrewniony z wirusem znalezionym u nietoperzy i łuskowców, wariant B pochodzi od A i różni się od niego dwiema mutacjami, a C pochodzi od B.
Autorzy badań informują, że zidentyfikowali dwie podgromady typu A, które różnią się od siebie mutacją synonimiczną (cichą). Wśród analizowanych danych do jednej z podgromad należały wirusy uzyskane od czterech Chińczyków z prowincji Guangdong, których wirus był oryginalnym wirusem podgromady oraz u trzech Japończyków i dwóch Amerykanów. W wypadku obu tych narodowości w genomie wirusa zaszły liczne zmiany. Wiadomo też, że obaj Amerykanie mieszkali w Wuhan. Z kolei do drugiej podgromady należały wirusy od pięciu osób z Wuhan (w tym w dwóch przypadkach była to oryginalna odmiana tej podgromady) oraz ośmiu innych osób z Chin i innych krajów Azji Wschodniej. Tutaj warto zauważyć, że aż 15 z 33 typów wirusów należących do tej podgromady znaleziono poza Azją Wschodnią, głównie w USA i Australii.
Jeśli zaś chodzi o koronawirusa SARS-CoV-2 typ B, to aż 74 z 93 wirusów z tej gromady znaleziono w Wuhan, innych regionach wschodnich Chin i niektórych sąsiednich krajach. Poza Chinami znaleziono jedynie 10 przykładów na występowanie typu B – w USA, Kanadzie, Meksyku, Francji, Niemczech i Meksyku. Typ B pochodzi od typu A i różni się od niego jedną mutacją synonimiczną oraz jedną niesynonimiczną. W przypadku typu B zauważono też pewien interesujący fakt. Otóż o ile w Azji Wschodniej nie zmutował, to w każdym przypadku, gdzie wykryto go poza tym regionem świata, zauważono mutacje. Zjawisko to można wyjaśnić albo specyficzną drogą zakażeń, albo też przyjmując założenie, że typ B zaadaptował się do populacji Azji Wschodniej i żeby poza nią wyjść, musi mutować.
Z kolei typ C pochodzi od typu B, od którego różni się jedną mutacją niesynonimiczną. Jest to typ „europejski”, który ma bardzo silną reprezentację we Francji, Włoszech, Szwecji, Anglii oraz w Kaliforni i Brazylii. Nie znaleziono go w Chinach, ale występuje w Hongkongu, Korei Południowej i na Tajwanie.
Od czasu przeprowadzenia powyższych badań uczeni poszerzyli liczbę analizowanych przypadków to 1001 genomów koronawirusa. Przygotowali już artykuł, który nie został jeszcze zrecenzowany, ale już informują, że na podstawie nowych badań można stwierdzić, że do pierwszej infekcji SARS-CoV-2 u ludzi doszło pomiędzy połową września a początkiem grudnia.
Potwierdza więc się to, o czym donosili dziennikarze z South China Morning Post. Widzieli oni tajne dokumenty rządowe, z których wynika, nie później niż 17 listopada nową chorobę zidentyfikowano u 9 osób.
Szczegóły badań opublikowano na łamach PNAS.


Komentarze (2)
Szkoda Mojego Czasu, 11 kwietnia 2020, 13:01
To badanie pokazuje jeszcze coś: że głosy Chińczyków, jakoby wirusa do Wuhan przywieźli amerykańscy marines, ktorzy tam byli na gościnnych wyjazdach nie sa wcale takie nieprawdopodobne. Typ A rozpoznano głównie u Amerykanów, a typ B (wuhański) jest jego klonem.
lanceortega, 13 kwietnia 2020, 21:01
Właśnie tak! Jest to prawdopodobne.
Rzeczywiście możliwe. Jedną z opcji.
Ale zachodnia autocenzura nie będzie o tym informowała. Na Zachodzie ludzie mają być powszechnie przekonani, że wirus pochodzi od prymitywnych chińskich wieśniaków jedzących wszelkie ścierwo - nawet nietoperze, pangoliny, żaby, pająki.....